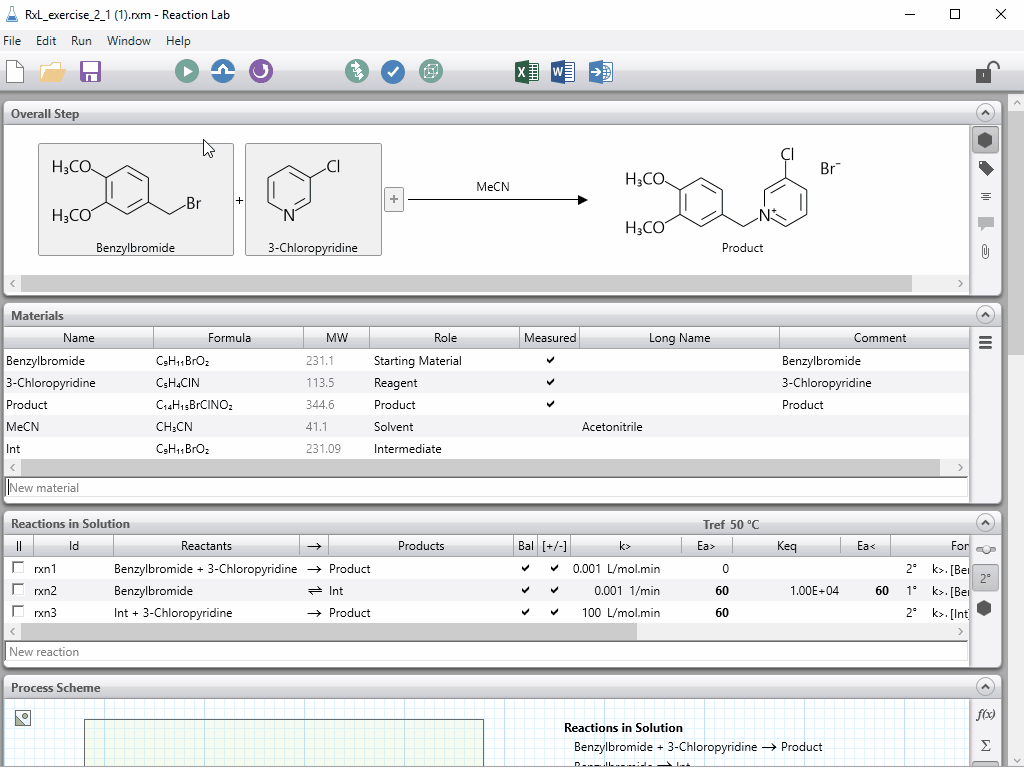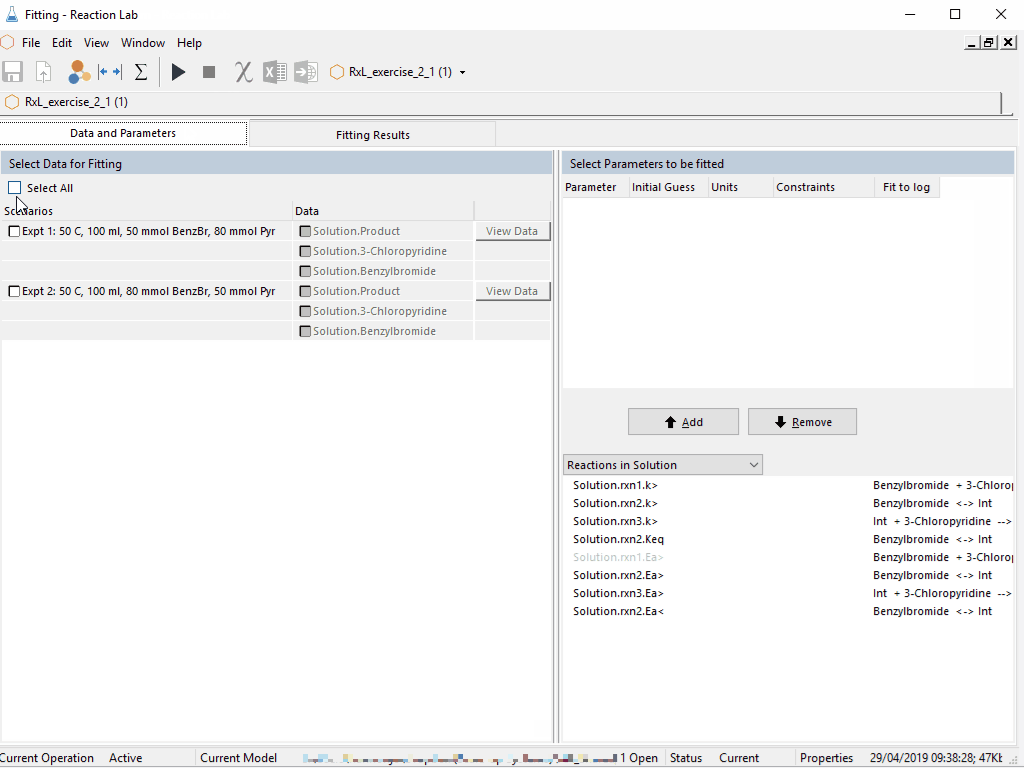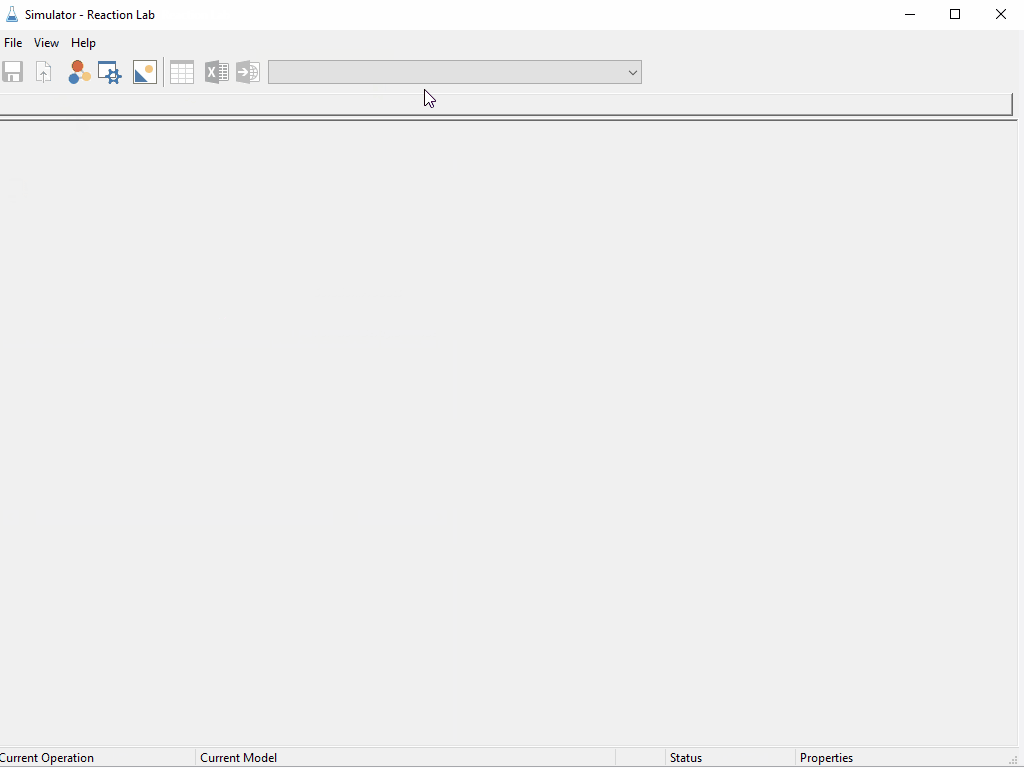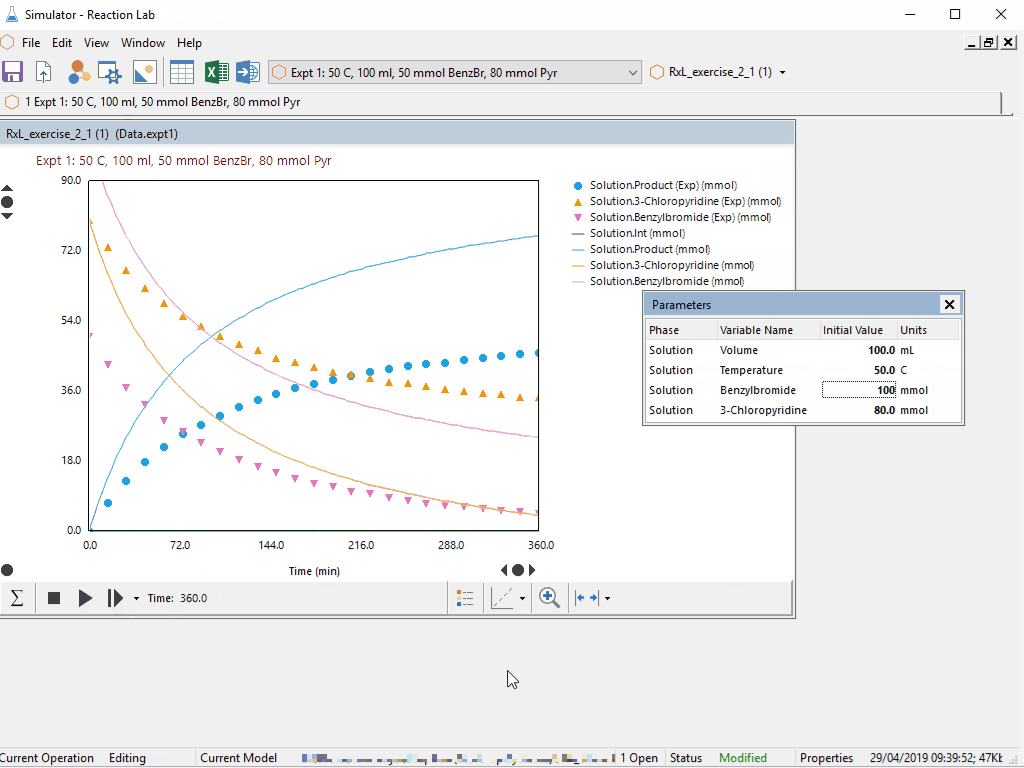
Last week, Peter Clark gave a preview of new features coming with Scale-up Suite 2. If you missed the event live, as usual you can catch the recoding in the resources site here.

Peter showed there is something for everyone in the new release. Whatever modality of drug/ active ingredient you develop or make, whether a small or large molecule or somewhere in between, whether made with cell culture or synthetic organic chemistry, your teams and your enterprise can obtain value daily from our tools. That takes us several steps closer to our vision to positively impact development of every potential medicine.
Scale-up Suite 2 includes:
- Powerful equipment calculators for scale-up, scale-down and tech transfer, leveraging our industry standard vessel database format
- Rigorous material properties calculators for pure components, mixtures and your proprietary molecules
- Empirical / machine learning tools, to build and use regression models from your data with just a few clicks; including support for DRSM
- Mechanistic modeling of any unit operation, in user-friendly authoring and model development environments
- Hybrid modeling, combining the best of both worlds
- Interactive data visualization, including parallel coordinates and animated contour plots for multidimensional datasets
- New features to make modeling faster, more productive and more enjoyable, incorporating ideas suggested by customers and from our own team
- New capabilities for autonomous creation of models, parameter fitting and process optimization 'headless' on the fly, as well as incorporation of real time data and access from any device.
- Interdisciplinary collaboration accelerates process development and innovation
- Models facilitate collaboration and knowledge exchange
- Interactive, real-time simulations save days and weeks of speculation
- Models are documents with a lifecycle extending from discovery to patient
- Model authoring tools must be convenient and easy to use
- Teams needs models that are easily shared
- Enterprises need tools that embed a modeling culture and support wide participation.
- an Online Library, containing hundreds of templates, documentation and self-paced training
- Free 1-hour on-line training events monthly
- Half-day and full day options for face to face training, available globally
- A free certification program to formally recognize your progress and skills
- Outstanding user support from PhD qualified experts with experience supporting hundreds of projects like yours
- A thriving user community, with round tables and regular customer presentations sharing knowledge and best practices.
We're celebrating 21 years serving the industry this year, supporting more than 20,000 user projects annually, for more than 100 customers all over the world, including 15 of the top 15 pharma companies.
If you're an industry, academic or regulatory practitioner, we invite you to join our user community and start to reap the benefits for your projects.